
13 Gel electrophoresis protocol
Gel Electrophoresis Protocol
Background Information adapted from Addgene
Gel electrophoresis is the standard lab procedure for separating DNA by size (e.g., length in base pairs) for visualization and purification. Electrophoresis uses an electrical field to move the negatively charged DNA through an agarose gel matrix toward a positive electrode. Shorter DNA fragments migrate through the gel more quickly than longer ones. Thus, you can determine the approximate length of a DNA fragment by running it on an agarose gel alongside a DNA ladder (a collection of DNA fragments of known lengths).
First, you need to decide what % agarose you will use. The greater the percentage of agarose, the smaller the linear DNA that can be resolved. The sugar polymers that make up the agarose gel matrix (powdered agarose heated in appropriate buffer, poured into a gel tray and allowed to solidify) act like a sieve. The greater the agarose concentration, the smaller the pores created in the gel matrix, and the more difficult (and slower) it is for large linear DNA molecules to move through the matrix.
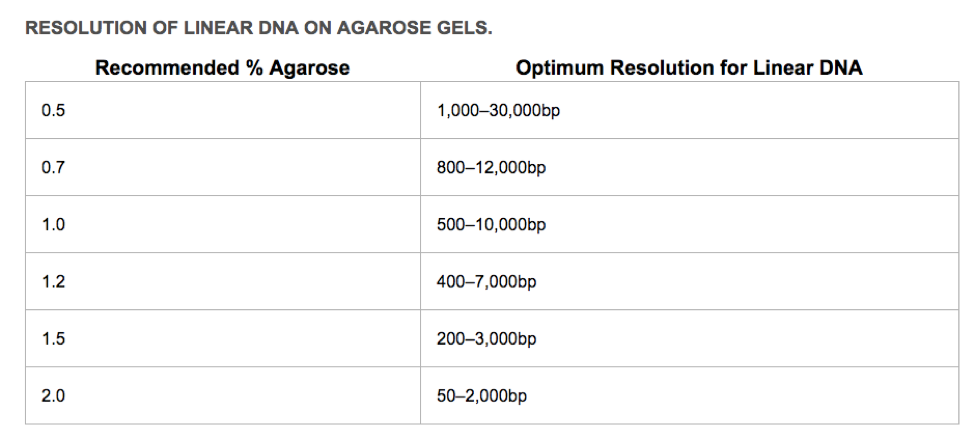
How to pour a 1% agarose gel (If pouring a different percentage gel, you must ADJUST your agarose values)
- Calculate how much agarose you need and measure it out on weigh paper.
For the Biorad mini-gel trays, gels need to be 30 mL in volume. For a 1% gel, you would weigh out 0.3 g agarose.
For the larger Carolina gel trays, gels need to be 50 mL in volume. For a 1% gel, you would weigh out 0.5 g agarose.
- Mix agarose powder with 1X Gel Running Buffer in an Erlenmeyer flask. Use 30 mL for the mini-gels, and 50 mL for the larger gels.
Two different running buffers are commonly used for DNA agarose gels. You will use the same running buffer to make up your gel and to submerge the gel in as you separate DNA fragments by size. Make sure you always dilute your running buffers to a 1X solution before using them.
1X Tris-acetate-EDTA (TAE) buffer contains: 40 mM Tris (pH 7.6), 20 mM acetic acid, and 1 mM EDTA. TAE is often prepared in concentrated stock solutions of 10× or 50×.
1X Tris-borate EDTA (TBE) buffer contains 100 mM Tris, 100 mM boric acid, and 2 mM EDTA. TBE is usually prepared as a 10X stock solution.
There are pros and cons to each type of running buffer: TBE is less prone to overheating, but some downstream enzymatic applications are inhibited by the borate in the running buffer. Migration is slower with TBE gels. TBE is more expensive than TAE, and cannot be prepared at as high of concentrations. But, TBE is better for resolving smaller DNA fragments.
- Microwave until the agarose is completely dissolved (but do not overboil the solution, as some of the buffer will evaporate and thus alter the final percentage of agarose in the gel. Many people prefer to microwave in pulses, swirling the flask occasionally as the solution heats up.)
Note: Caution: this solution will be HOT! Be careful while swirling, since eruptive boiling can occur. It is a good idea to microwave until it is almost boiling, stop and swirl, and then continue towards a boil. Keep an eye on it at all times, since it has a tendency to boil over.
- Let agarose solution cool down to about 50 °C (about when you can keep your hand on the flask), which takes about 3 mins.
- During this time, prepare your casting tray by taping the ends with masking tape. Place a comb in the tray, and make sure you have enough wells for all of your samples.
- Once your agarose has cooled slightly, add GelRed DNA Stain and swirl to mix. GelRed binds to the DNA and allows you to visualize the DNA under ultraviolet (UV) light. Our GelRed stock is at a concentration of 25,000X, so you must dilute it in your agarose mixture to a final concentration of ~1X (use 2 μl of stock solution per 50 mL gel).
- Pour the agarose into a gel tray with the comb in place. Note: Pour slowly to avoid bubbles which will disrupt the gel. Any bubbles can be pushed away from the well comb or towards the sides/edges of the gel with a pipette tip.
- Let gel sit at room temperature for ~20 min, until it has completely solidified.
Note: If you are in a hurry the gel can also be set more quickly if you place the gel tray at 4°C earlier so that it is already cold when the gel is poured into it.
Loading Samples and Running an Agarose Gel:
- Add loading buffer to all samples. Note: some PCR buffers already contain loading dye—check the manufacturer’s guidelines!
Note: Loading buffer serves two purposes: 1) it provides a visible dye that helps with gel loading and will also allow you to gauge how far the gel has run while you are running your gel; and 2) it contains a high percentage of glycerol, so it increases the density of your DNA sample causing it settle to the bottom of the gel well, instead of diffusing in the buffer.
- Once solidified, remove the tape and comb from your gel. The gel stays in its plastic tray. Place the agarose gel (on its tray) into the gel box (the electrophoresis unit).
- Fill gel box with 1X running buffer until the gel is just covered. The surface should look entirely smooth. Use the same running buffer you used to prep the gel.
- Carefully load a DNA ladder into the first lane of the gel. If you won’t use all the lanes, the goal is to ‘center’ your samples. Note: When loading the sample in the well, place the very top of the tip of the pipette into the buffer just above the well. Very slowly and steadily, push the sample out and watch as the sample fills the well. After all of the sample is unloaded, push the pipettor to the second stop and carefully raising the pipette straight out of the buffer.
- Carefully load your samples into the additional wells of the gel.
- Run the gel at 80-100 V until the dye line is approximately 75-80% of the way down the gel. Note: Black is negative, red is positive. (The DNA is negatively charged and will run towards the positive electrode.) Always Run to Red.
- Turn OFF the power, disconnect the electrodes from the power source, and then carefully remove the gel from the gel box.
- Using a device that has UV light, visualize your DNA fragments.
Note: When using UV light, protect your skin by wearing safety goggles or a face shield, gloves and a lab coat.
Note: The fragments of DNA are usually referred to as ‘bands’ due to their appearance on the gel.
Analyzing Your Gel:
- Using the DNA ladder in the first lane as a guide, you can interpret the bands that you get in your sample lanes to determine if the resulting DNA bands that you see are as expected or not.
- Label the bands of your ladder and label the sizes of resulting amplicons.
Ladders for DNA gels
NEB 100 bp DNA ladder: Use for amplicons LESS THAN 1000 bp
Protocol for DNA ladder: Use 1 uL of ladder stock per lane. To dilute the ladder for loading, mix 1 uL of stock ladder with 1 uL 6X DNA loading dye and 4 uL of water. Load the entire 6 uL volume into one well.
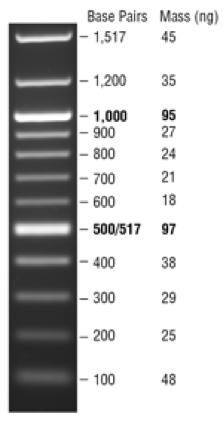
NEB 1 kb Plus DNA Ladder for Safe Stains: Use for amplicons > 1000 bp:
Load 5 µl (0.25 µg) per gel lane
Note: this ladder already has loading dye, so do not add more—just load the 5 ul.
