
7 SNP Independent Project
BIOL 344 Independent Research Projects
Objective: Develop a PCR-based genotyping assay to investigate a biological question of your choice. You will:
- Work with your partner to choose a SNP associated with a phenotype that you are interested in. Previous students have evaluated the genetic basis of anxiety, taste ability, eye color, dyslexia, ability to digest lactose, and more!
You will be designing a PCR assay to distinguish DNA sequences that differ by a single nucleotide. Try to identify a SNP where you have a high chance of finding both SNP alleles in our population: if you pick a very rare mutation, it is unlikely that you will find anyone with the rare mutation, and thus you won’t be able to tell if your PCR assay is working. So how can you find a SNP of interest?
- Here are the traits the company 23andme investigates in their genetic analysis. You may need to do some googling/searching in the scientific literature to find the identifier for the SNP they are referring to. SNPs have unique identifiers that begin with the letters “rs” and then have a long string of numbers.
- You can search scientific literature (Web of Science, PubMed, or Google scholar) for the word SNP and a disease/trait you care about to identify known associations
- Encyclopedia of fun and interesting SNPs: https://www.snpedia.com/
- One you have the SNP identifier (the rs number), use the NCBI SNP data to get more info on allele frequency: https://www.ncbi.nlm.nih.gov/snp/
- Identify the gene involved in this disease/phenotype (e.g.CFTR, beta globin, dystrophin, BRCA1, p53). The SNP you have chosen may either be located within the gene, or in a noncoding region surrounding a gene, where it could influence gene expression. One good place to research the genetic basis of human diseases/phenotypes is here: https://www.omim.org/.
- We will design primers to try to distinguish the genotypes of you and your peers. You will also collect data on the phenotypes of your test subjects. To learn about their phenotypes, you could give a survey or make observations. Your goal will be to see if you can make a link between SNP genotypes and phenotypes.
If you have another idea that doesn’t fit this basic strategy, talk to me. Creativity is always welcome.
In lab, you will work with your partner to decide on your target and design primers to amplify your SNP. You will ultimately have to agree on one SNP, but having a couple options is important because some areas of the genome do not work well for primer design. With your partner, you will work together to decide: what is your research goal? Which SNP are you going to focus on? What phenotype are you studying? How will you collect phenotype data?
Primer design plan
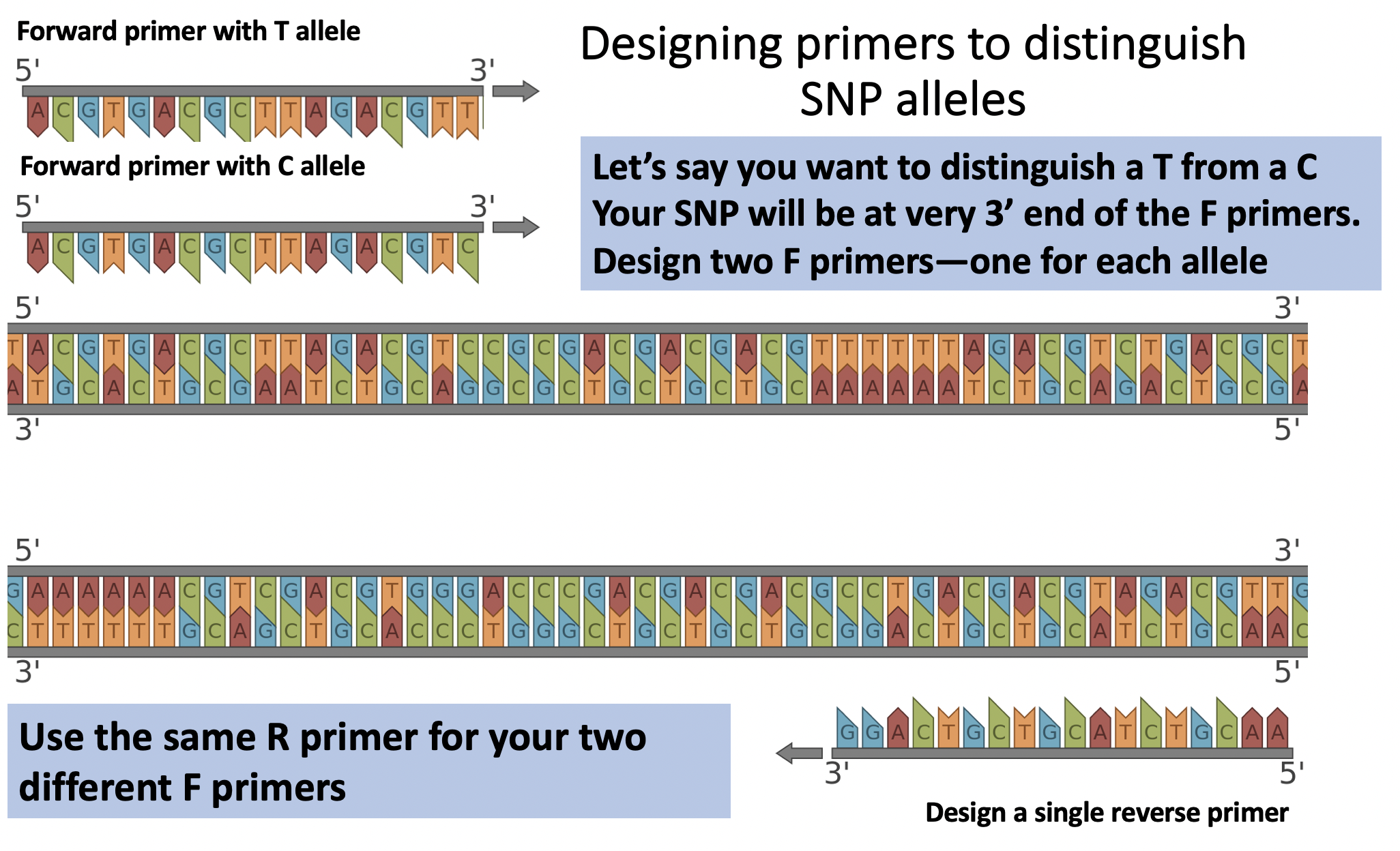
Once you’ve identified your SNP of interest, you’ll design PCR primers that distinguish between two nucleotide options (two possible alleles) at your SNP. You’ll need to design two different forward primers (one for each SNP allele) and a single reverse primer that binds downstream from your SNP of interest. We want to design forward primers so that they will ONLY enable amplification if a specific SNP is present in someone’s genome.
Yaku method of primer design
Method adapted from a lab developed by Vincent Cracolici
To more reliably use PCR to distinguish our two SNP alleles, we’ll be using a special primer design technique called the Yaku-Bonczyk method. The Yaku method can increase the likelihood that a F primer ONLY enables amplification when a specific SNP allele of interest is present; this technique thus reduces our ‘false positive’ rate. How does it work? The standard method of primer design for a genetic mutation (shown in the figure above) involves two forward primers which are identical except for the last nucleotide at the 3’ end: one primer is complementary to wild-type DNA (one allele) and the other to mutant DNA (the other possible SNP allele). However, the single base-pair mismatch between these primers and non-target DNA is often not enough to ensure that the wild-type primer will not anneal to and extend mutant DNA, and vice versa. The Yaku-Bonczyk method differs from standard PCR methods because the primers are designed to better discriminate against non-complementary DNA by incorporating an intentional mismatch into the primer. The Yaku-Bonczyk method requires the most 3’ nucleotide of each forward primer to be complementary to the mutant/wild-type DNA it is seeking, the second nucleotide in is designed to always anneal to the template DNA, and the third nucleotide in is designed as an intentional mismatch that will never anneal to either wildtype or mutant DNA (see illustration below). By slightly altering the 3’ end of our wild-type and mutant primers, we can drastically increase primer discrimination against nonspecific binding. If a primer and its complementary DNA strand anneal to each other, the single mismatch three base pairs in from the 3’ end is not enough to prevent extension and will result in a band indicating successful amplification. A primer designed with the Yaku-Bonczyk method should not anneal to nor extend non-target DNA as a result of the two mismatches. The attraction at the second base pair in on the primer is not enough to allow for extension at the 3’ end. Therefore, nonspecific binding is decreased and our primers should distinguish the two SNP alleles.
F Primer example: 5’ TCGCACTGCGAACGTGGTCXYZ 3’
Things to consider when choosing your primers:
- Make your primers ~20-28 nt long if possible.
- Your primer melting temps must be compatible (within 3-5 degrees C of each other). Aim for about 62 degrees C if possible.
- GC content should be ~50% if possible (in Benchling if you drag across a section of DNA, it will tell you the GC content, melting temp, and length, which can be useful in figuring out how long to make your primer).
- Since you are introducing a different nucleotide into position X (3 nucleotides in from the end), you can choose a nucleotide that yields the most desirable primer properties (GC content and Tm)

Once you’ve designed your two F primers, you’ll design a single R primer that will be used to amplify a segment of your target DNA. The exact position of the R primer is not important–the F primer is how we distinguish the two SNP alleles. You do NOT use the Yaku method when designing the R primer, since it is NOT distinguishing between different sequences. You’ll choose a R primer that yields a PCR amplicon between 200 bp – 1000 bp. Aim for ~500 bp if possible so we can utilize similar PCR extension times across groups.
Interpreting genotype from your gel results
This technique allows you to use PCR amplification and gel electrophoresis as your readout for whether each SNP allele is present in someone’s genome. Don’t forget that as diploid organisms, we have two copies of each autosomal (non-sex chromosome) gene. Therefore, your participants may possess both SNP alleles (heterozygous) or have two of the same SNP allele (homozygous). You’ll determine someone’s genotype by separately analyzing if they possess each of the two alleles for your SNP of interest. The presence or absence of a band indicates allele presence, as shown in the gels below assaying if ten different participants have a G or A (or both) at one particular SNP. Since our two F primers bind to the same position in the genome and we use the same R primer, the amplicon size is the same regardless of which F primer was used. Once the presence or absence of a band is recorded, you can determine the genotype of each participant. For example, in the gel below, you can find participants who are AA, GA, and GG at this SNP.
